445822357
- жµПиІИ: 742686 жђ°
-

з§ЊеМЇзЙИеЭЧ
- жИСзЪДиµДиЃѓ ( 0)
- жИСзЪДиЃЇеЭЫ ( 0)
- жИСзЪДйЧЃз≠Ф ( 0)
е≠Шж°£еИЖз±ї
- 2014-10 ( 19)
- 2014-09 ( 18)
- 2014-08 ( 18)
- жЫіе§Ъе≠Шж°£...
жЬАжЦ∞иѓДиЃЇ
-
dfjjfxylпЉЪ
еЉАжЇРй°єзЫЃжО®иНРзљСзЂЩ:http://binlily.imwork. ...
JAVAеЉАжЇРй°єзЫЃ -
еЦµеЦµе§Із•ЮпЉЪ
ињЩз±їеЕНиієAPIињШжШѓжМЇе§ЪзЪДпЉМеНЪеЃҐдЄКдєЯжХізРЖињЗпЉЪhttps://my ...
Web Api --жЩЇиГљApiжО•еП£
еЕ®еЯЇеЫ†зїДйЗНжµЛеЇП

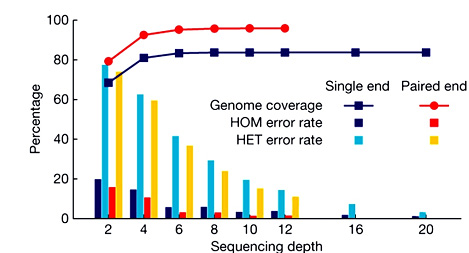
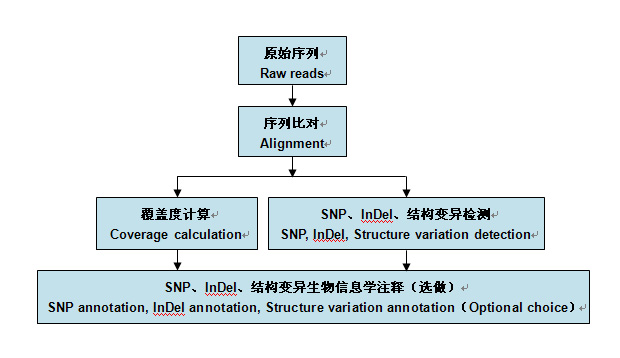
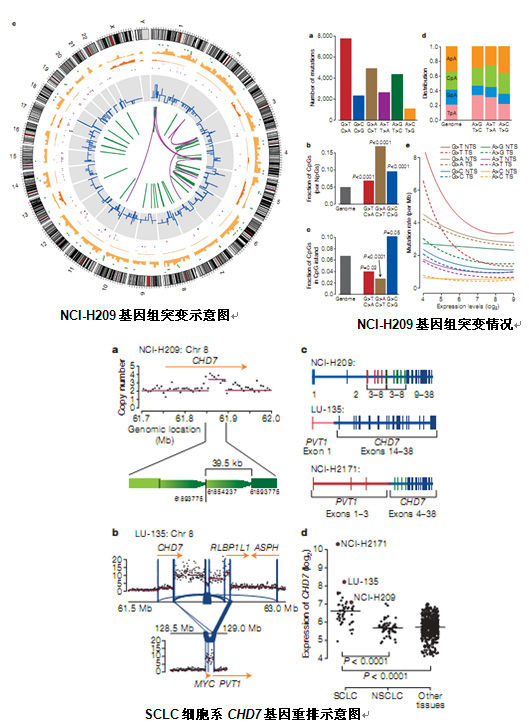



зЫЄеЕ≥жО®иНР
еЕ®еЯЇеЫ†зїДйЗНжµЛеЇПжХ∞жНЃеИЖжЮР.doc
еЕ®еЯЇеЫ†зїДйЗНжµЛеЇПжХ∞жНЃеИЖжЮРжК•еСК.doc
Ion torrentеЊЃзФЯзЙ©(зїЖиПМ)еЕ®еЯЇеЫ†зїДйЗНжµЛеЇПжЦЗеЇУжЮДеїЇеЃЮй™МжЦєж°И.pdf
IontorrentеЊЃзФЯзЙ©(зїЖиПМ)еЕ®еЯЇеЫ†зїДйЗНжµЛеЇПжЦЗеЇУжЮДеїЇеЃЮй™МжЦєж°И.pdf
еН±йЗНжЦ∞зФЯеДњйБЧдЉ†жАІзЦЊзЧЕењЂйАЯеЕ®еЯЇеЫ†зїДжµЛеЇПдЄУеЃґеЕ±иѓЖ
2020-еЃПеЯЇеЫ†зїДе≠¶жµЛеЇПжКАжЬѓеЬ®дЄ≠йЗНзЧЗжДЯжЯУдЄ≠зЪДдЄіеЇКеЇФзФ®дЄУеЃґеЕ±иѓЖ.pdf
еЯЇеЫ†зїДдЇМдї£жµЛеЇПжХ∞жНЃзЪДиЗ™еК®еМЦеИЖжЮРжµБз®ЛеЯЇеЫ†зїДдЇМдї£жµЛеЇПжХ∞жНЃзЪДиЗ™еК®еМЦеИЖжЮРжµБз®Л
ж∞із®їз±љз≤ТдљОйХЙеУБз≥їзЪДеЯЇеЫ†зїДйЗНжµЛеЇПеИЖжЮРпЉМзОЛеЃЗиОєпЉМиВЦзВОеЗ§пЉМж∞із®їжШѓжИСеЫљжЬАдЄЇйЗНи¶БзЪДз¶Њи∞Јз±їдљЬзЙ©дєЛдЄАпЉМињСеєійЪПзЭАзОѓеҐГж±°жЯУзЪДеК†еЙІпЉМж∞із®їеПЧйЗНйЗСе±ЮйХЙж±°жЯУзЪДжГЕеЖµжЧ•зЫКдЄ•йЗНгАВеРМжЧґпЉМж±°жЯУж∞із®їзЪДйХЙдЉЪзїПзФ±й£Я
жѓФиЊГиѓ¶зїЖеЬ∞дїЛзїНеЯЇеЫ†зїДжµЛеЇПжµБз®Л
иЗ™еК®еМЦеНХзЂѓеЕ®еЯЇеЫ†зїДйЗНжµЛеЇП (WGRS) жХ∞жНЃе§ДзРЖпЉМдїОиАМдљњзФ®йҐДеЃЙи£ЕзЪДдЊЭиµЦй°єе∞ЖиѓїеПЦдїО FASTQ жШ†е∞ДеИ∞еПВиАГеєґйЗНжЦ∞еѓєйљРжПТеŕ犯姱гАВ BWA ењЕй°їеЃЙи£ЕеєґеЬ®з≥їзїЯиЈѓеЊДдЄКеПѓзФ®пЉМиАМ GenomeAnalysisTK.jar ењЕй°їеЬ® MATLAB иЈѓеЊДдЄКеПѓзФ®гАВ е¶ВжЮЬ...
дЄ≠еЫљеЃПеЯЇеЫ†зїДе≠¶зђђдЇМдї£жµЛеЇПжКАжЬѓж£АжµЛжДЯжЯУзЧЕеОЯдљУдЄіеЇКеЇФзФ®дЄУеЃґеЕ±иѓЖ2020
еЯЇеЫ†зїДеТМиљђељХзїДйЂШйАЪйЗПжµЛеЇПжХ∞жНЃеИЖжЮРжµБз®ЛеТМеИЖжЮРеє≥еП∞
40дЄ™еЯЇеЫ†зїДеЃМеЕ®йЗНжµЛеЇПжП≠з§ЇиЪХзЪДй©ѓеМЦдЇЛдїґеПКеЕґзЫЄеЕ≥еЯЇеЫ†[жФґйЫЖ].pdf
дЇЇз±їеЯЇеЫ†зїДжµЛеЇПжЦЗжЬђжХ∞жНЃжМЦжОШз†Фз©ґ.pdf
еЊЃзФЯзЙ©еЯЇеЫ†зїДжµЛеЇПеЬ®ељУдїКзФЯеСљзІСе≠¶йҐЖеЯЯзЪДз†Фз©ґдЄ≠иµЈзЭАйЗНи¶БдљЬзФ®гАВжЬђжЦЗйАЪињЗеѓєдЄїи¶БжµЛеЇПжЦєж≥ХдЄАйЄЯжЮ™ж≥ХзЪДдїЛзїН,йШРжШОдЇЖеЊЃзФЯзЙ©еЕ®еЯЇеЫ†зїДеЇПеИЧзЪДжµЛеЃЪдЄОж≥®йЗКеЬ®еЯЇеЫ†зїДе≠¶зЪДињЫдЄАж≠•з†Фз©ґдЄ≠зЪДеєњйШФйҐЖеЯЯгАВ
еЯЇдЇОеЕ®еЯЇеЫ†зїДеЕ≥иБФеИЖжЮРзЪДе∞ПйЇ¶дЇІйЗПйБЧдЉ†зїУжЮДиІ£жЮР.pdf
еЕ≥дЇОйЂШйАЪйЗПжµЛеЇПеЬ®дЄ™жАІеМЦж≤їзЦЧеПКз≤ЊеЗЖеМїзЦЧдЄ≠зЪДдљЬзФ®
дЄЙдї£еЯЇеЫ†зїДжµЛеЇПжКАжЬѓзЃАдїЛеПКеЕґеОЯзРЖжХізРЖзђђдЄАдї£жµЛеЇПжКАжЬѓзђђдЄАдї£DNAжµЛеЇПжКАжЬѓзФ®зЪДжШѓ1975еєізФ±ж°Сж†ЉпЉИSangerпЉЙеТМиАГе∞Фж£ЃпЉИCoulsonпЉЙеЉАеИЫзЪДйУЊзїИж≠Ґж≥Хдї•еПК1976-1977еєізФ±й©ђеЕЛи•њеІЖпЉИMaxamпЉЙеТМеРЙе∞Ф
зУ¶еРЙиѓЇзФ®дЇОеЕ®еЯЇеЫ†зїДжµЛеЇПжХ∞жНЃеТМе§ІеЮЛSNPжХ∞жНЃеЇУзЪДFase SNPеЯЇеЫ†еИЖеЮЛеЈ•еЕЈгАВдїОBiocondaеЃЙи£ЕеПѓдї•дљњзФ®еСљдї§conda install vargenoдїОBioconda conda install vargeno гАВ иѓЈиљђеИ∞дї•иОЈеПЦжЬЙеЕ≥BiocondaзЪДжЫіе§Ъдњ°жБѓгАВ е¶ВжЮЬжВ®е∞ЪжЬ™ пЉМеИЩ...